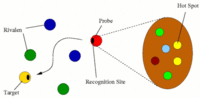
Transport of nutrients to peripheral tissues and healing of damaged blood vessels are among the most important functions of blood. These functions involve the action of a series of proteins some of which are found in large amounts in the blood circulation. Fibrinogen is a multiprotein complex which, when activated, aggregate to form fibrin, a net-shaped molecular formation which is fundamental for the coagulation of blood following, i.e, a wound or when an extraneous body comes into contact with blood (i.e., graft implants). Thus, adsorption of fibrinogen on material surfaces play an important role in viability of those materials for implants. Transport of nutrients to peripheral tissues and healing of damaged blood vessels are among the most important functions of blood. These functions involve the action of a series of proteins some of which are found in large amounts in the blood circulation. Fibrinogen is a multiprotein complex which, when activated, aggregate to form fibrin, a net-shaped molecular formation which is fundamental for the coagulation of blood following, i.e, a wound or when an extraneous body comes into contact with blood (i.e., graft implants). Thus, adsorption of fibrinogen on material surfaces play an important role in viability of those materials for implants.
In collaboration with experimental groups in the field, we use atomistic molecular dynamics simulations to characterize the adsorption process of fibrinogen on material surfaces. Another important molecule in the blood is albumin, which mediate transport of lipids and other molecules in blood. Albumin is a multidomain protein which provides several binding sites used to bind a range of different target molecules. Target molecules (lipids, drugs, etc.) bind to albumin which act as a transporter, and are then released where needed by blood circulation. Here we use molecular dynamics simulations to study the binding modes of several lipids to Albumin and the kinetics of lipid release/uptake. If you are interested, please contact Friederike Schmid or Giovanni Settanni.
|
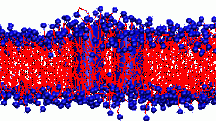 All living things depend on membranes. Their basic structure is provided by lipid bilayers, which self-assemble spontaneously in water due to the amphiphilic character of lipid molecules - they contain both hydrophilic and hydrophobic units. In our group, we are interested in generic properties of such amphiphilic bilayers.
All living things depend on membranes. Their basic structure is provided by lipid bilayers, which self-assemble spontaneously in water due to the amphiphilic character of lipid molecules - they contain both hydrophilic and hydrophobic units. In our group, we are interested in generic properties of such amphiphilic bilayers.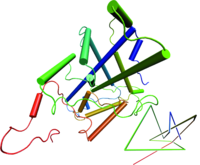 Although globular homopolymers are typically highly knotted, less than one in a hundred protein structures contain a knot ( http://knots.mit.edu ). Nevertheless, intriguing counter-examples exist, like the most complicated protein knot, which was discovered recently during a diploma thesis in our group (see figure on the left). Apart from analyzing biological data, we perform Monte Carlo simulations of simplified protein and DNA models to learn more about entanglements in viral DNA, chromatin and proteins. On this topic, we collaborate with theory groups at MIT and an experimental group at the MPI for Polymer Research. If you are interested in interdisciplinary investigations at the frontier of physics, mathematics and molecular biology, please contact
Although globular homopolymers are typically highly knotted, less than one in a hundred protein structures contain a knot ( http://knots.mit.edu ). Nevertheless, intriguing counter-examples exist, like the most complicated protein knot, which was discovered recently during a diploma thesis in our group (see figure on the left). Apart from analyzing biological data, we perform Monte Carlo simulations of simplified protein and DNA models to learn more about entanglements in viral DNA, chromatin and proteins. On this topic, we collaborate with theory groups at MIT and an experimental group at the MPI for Polymer Research. If you are interested in interdisciplinary investigations at the frontier of physics, mathematics and molecular biology, please contact 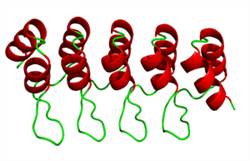 Repeat protein domains are formed by tandem arrays of repeating structural units, constitute about 20% of the eukaryotic proteome, mediating protein-protein interactions and acting as mechano-transductors. As such they may represent the basis for the construction of mechanical nanodevices. In collaboration with experimental groups in the field, we have been working on simplified models of repeat proteins which explains both the thermodynamics and the kinetics of folding of this class of proteins. We have also been carrying out atomistic molecular dynamics (MD) simulations of several repeat protein systems to study their folding behavior and their mechanical characteristics when subjected to external pulling forces. If you are interested, please contact
Repeat protein domains are formed by tandem arrays of repeating structural units, constitute about 20% of the eukaryotic proteome, mediating protein-protein interactions and acting as mechano-transductors. As such they may represent the basis for the construction of mechanical nanodevices. In collaboration with experimental groups in the field, we have been working on simplified models of repeat proteins which explains both the thermodynamics and the kinetics of folding of this class of proteins. We have also been carrying out atomistic molecular dynamics (MD) simulations of several repeat protein systems to study their folding behavior and their mechanical characteristics when subjected to external pulling forces. If you are interested, please contact 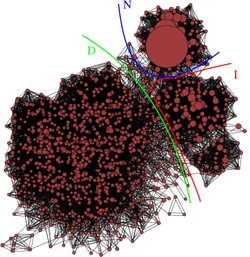 Our activity focuses on the development and application of methods for the identification of the folding transition state of peptides and, more in general, for the complete characterization and representation of the dynamics of peptides by using atomistic molecular dynamics simulations . This research effort is based on the application of concepts like kinetic networks (figure on the left) and Markov models to the trajectory data of peptides collected by MD simulations. Results from this line of research are validated against available experimental data on the kinetics of folding of peptides (folding/unfolding rates, phi values). If you are interested, please contact
Our activity focuses on the development and application of methods for the identification of the folding transition state of peptides and, more in general, for the complete characterization and representation of the dynamics of peptides by using atomistic molecular dynamics simulations . This research effort is based on the application of concepts like kinetic networks (figure on the left) and Markov models to the trajectory data of peptides collected by MD simulations. Results from this line of research are validated against available experimental data on the kinetics of folding of peptides (folding/unfolding rates, phi values). If you are interested, please contact